自A. W. Hofmann于1855年首次使用“芳香性”一词以来,芳香性成为化学中影响最广泛、最富活力的概念之一,而芳香化合物则在化学、生命和材料科学中扮演重要角色。在诸多描述芳香性的理论中,最经典的是Hückel规则,即(4n+2)个π电子为芳香性,而(4n)个π电子为反芳香性。在1972年,Baird提出三重态的芳香性判断规则,恰与Hückel规则相反。由于具有三重态基态的分子很罕见,Baird规则主要适用于激发态,而基态Baird芳香性的例子极少。8π电子的苯二负离子是研究芳香性的绝佳案例:根据Hückel规则,单重态苯二负离子呈现反芳香性;而Baird规则则预测三重态的苯二负离子应具有芳香性。但是,苯二负离子极易发生Jahn–Teller畸变,导致基态从D6h下的三重态变为D2h下的单重态(醌式或反醌式结构)(图1)。因此,目前文献中已报道的苯二负离子的基态均为单重态,而三重态基态的苯二负离子仍是空白。近日,77779193永利集团的黄闻亮课题组与高松课题组合作,利用稀土离子4f电子与苯二负离子之间的磁交换作用,首次实现了三重态基态的苯二负离子的稳定,并证实其具有Baird芳香性。该成果以“Synthesis and Stabilization of a Benzene Dianion with a Triplet Ground State and Baird Aromaticity”为题,于2025年2月21日在《美国化学会志》(Journal of the American Chemical Society)上在线发表。
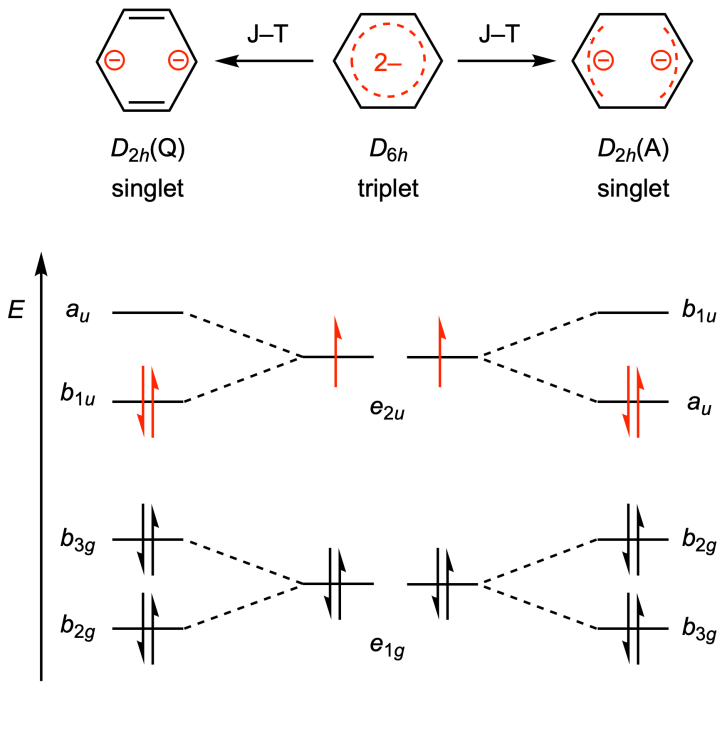
图1. Jahn–Teller效应与苯二负离子的基态
在黄闻亮团队前期合成中性的反三明治型稀土苯四负配合物的基础上(Chem. Sci. 2024, 15, 8740–8749),在该工作中,利用大位阻的β-二酮亚胺(BDI)合成了铕(II)和镱(II)的苯二负配合物[(BDI)M(THF)]2(μ-η6,η6-C6H6) (M=Eu: 2-Eu; M=Yb: 2-Yb)和[(BDI)M(THF)]2(μ-η6,η6-C6D6) (M=Eu:2-Eu-d6; M=Yb: 2-Yb-d6) (图2A)。核磁共振氢谱显示,2-Yb中苯二负离子的氢位于高场‒18 ppm处,而2-Yb-d6的氘谱也在‒16 ppm处观测到了相应的信号,表明2-Yb中苯二负离子可能存在反芳香性。此外,用对二甲苯替换苯,可以得到局域对称性降低的铕(II)的对二甲苯配合物[(BDI)Eu(THF)]2(μ-η6,η6-1,4-Me2C6H4) (3-Eu)。以上化合物均得到了单晶结构的表征,2-Eu-d6的结构如图2B所示。在这些配合物中,配位芳环的平均C‒C键长(1.42‒1.43Å)介于中性苯(1.391Å)与苯四负离子(1.44‒1.47Å)之间,与文献中苯二负离子的平均C‒C键长(1.42‒1.44Å)相近。此外,配位芳环保持高度的平面性(图2C),这与文献中大部分存在显著去平面化的苯二负离子不同。值得注意的是,在2-Yb-d6和3-Eu中,苯二负离子的C−C键长呈现出反醌式的“两长四短”模式,而在2-Eu-d6中,苯二负离子的C−C键长分布则较为均匀。以上结构特征表明,苯二负离子的电子结构可能同时受到稀土离子和局域对称性的影响。
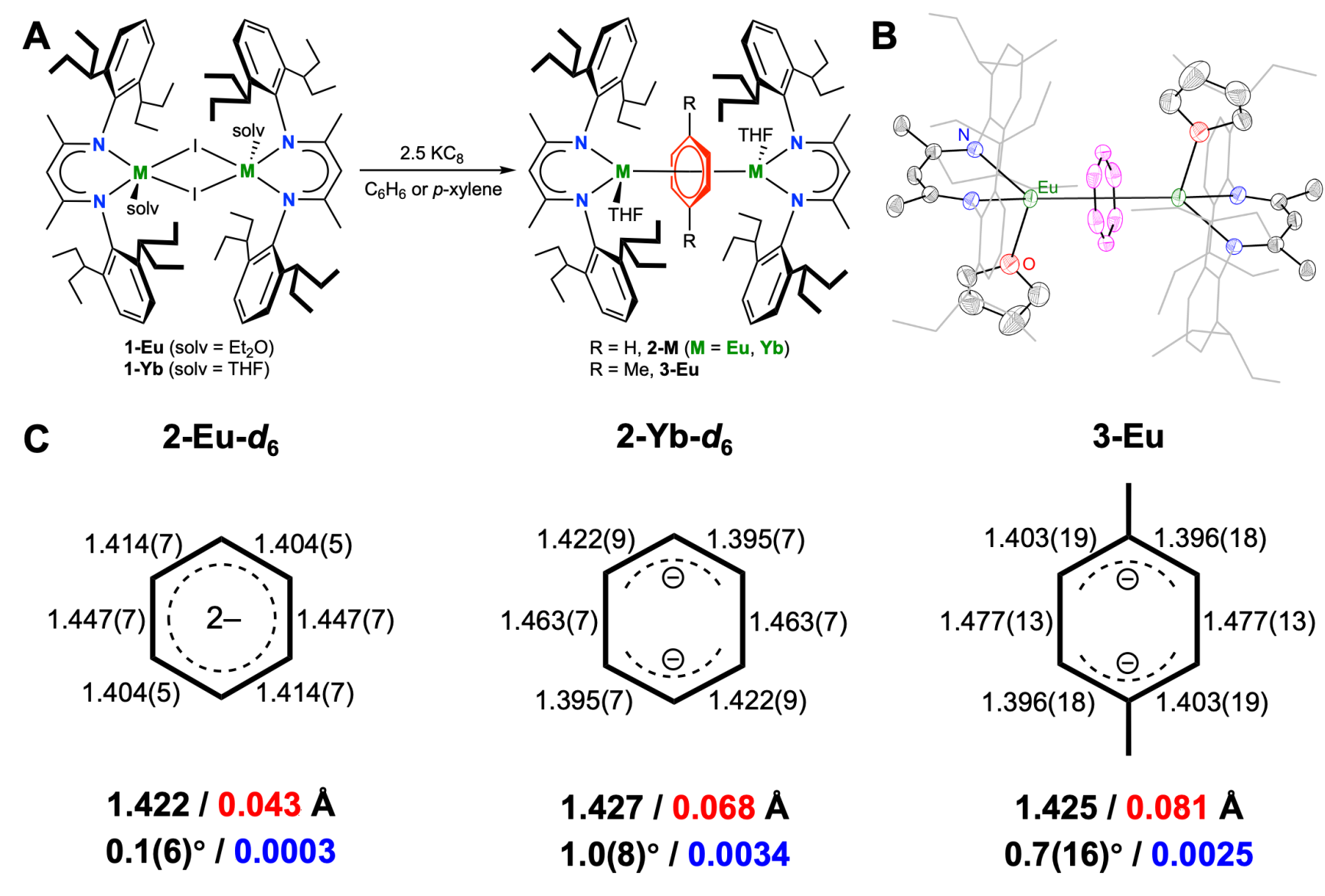
图2.铕(II)和镱(II)的苯二负配合物的合成与单晶结构
作者对这些配合物进行了全面的谱学和磁性表征来探究其电子结构。2-Yb、2-Eu和3-Eu戊烷溶液的吸收光谱均在500和700nm处有两个宽吸收带(ε~2000‒5000 M–1·cm–1)。EPR测试结果显示,2-Yb的基态为单重态,且存在能量上很接近的三重态激发态。磁性测试结果显示,在室温时,2-Eu的χmT值小于1-Eu和3-Eu,而后两者则相当于两个未耦合的Eu2+离子之和(图3A);在2 K时,2-Eu的磁化强度与两个Eu2+离子减去一个三重态苯二负离子的值相吻合(图3B)。若将2-Eu视为三中心自旋体系,用海森堡模型拟合得到苯二负离子与Eu2+间的磁耦合常数JEu–rad为–80 cm–1(图3C),而将2-Eu视作四中心自旋体系得到的JEu–rad相似且苯二负离子上的两个单电子呈现极强的铁磁耦合(J=510 cm–1),进一步证实了苯二负离子为三重态。与之相反,3-Eu在2 K下的磁化强度与两个未耦合的Eu2+离子相符,而对其磁化率进行拟合发现两个Eu2+之间仅存在很弱的反铁磁耦合。以上结果表明,在2-Eu中,苯二负离子具有三重态基态,且与两个Eu2+离子以反铁磁耦合,而在2-Yb和3-Eu中,苯二负离子均为单重态基态。值得指出的是,2-Eu中三重态苯二负离子与Eu2+间的磁耦合常数JEu–rad超过了文献中迄今报道的镧系离子与有机配体作用的最大值,而该强反铁磁耦合正是稳定苯二负离子三重态的关键。
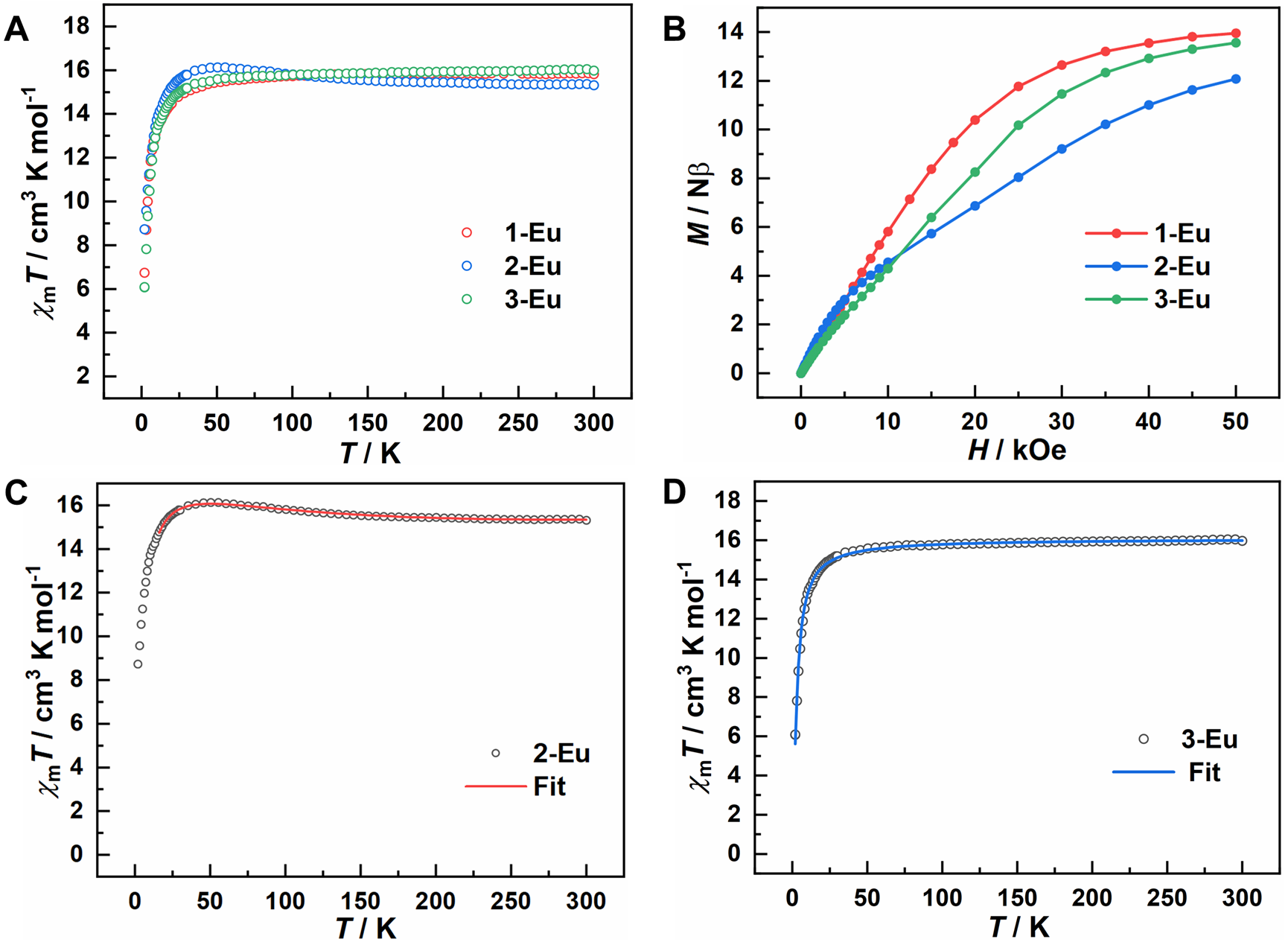
图3. 1-Eu、2-Eu和3-Eu的磁性数据(A)变温磁化率;(B)在2 K下的磁化强度;(C)2-Eu基于三中心自旋体系的拟合;(D)3-Eu基于两中心自旋体系的拟合
为了进一步探究电子结构,作者进行了密度泛函理论(DFT)的计算。对于2-Eu,计算结果表明13A态的能量最低,恰对应与Eu2+离子反铁磁耦合的三重态苯二负离子(图4A)。而3-Eu的15A的能量最低,表明对二甲苯二负离子的基态为单重态。对于2-Yb,基于单晶结构的计算结果表明开壳层单重态(OST)的能量最低,对应单重态苯二负离子,这也与EPR测试结果一致(图4B)。成键分析表明,2-Eu(13A)的两个近简并的单占据轨道(SOMOs)反映了Eu2+与苯二负离子之间的δ成键作用(图4C)。
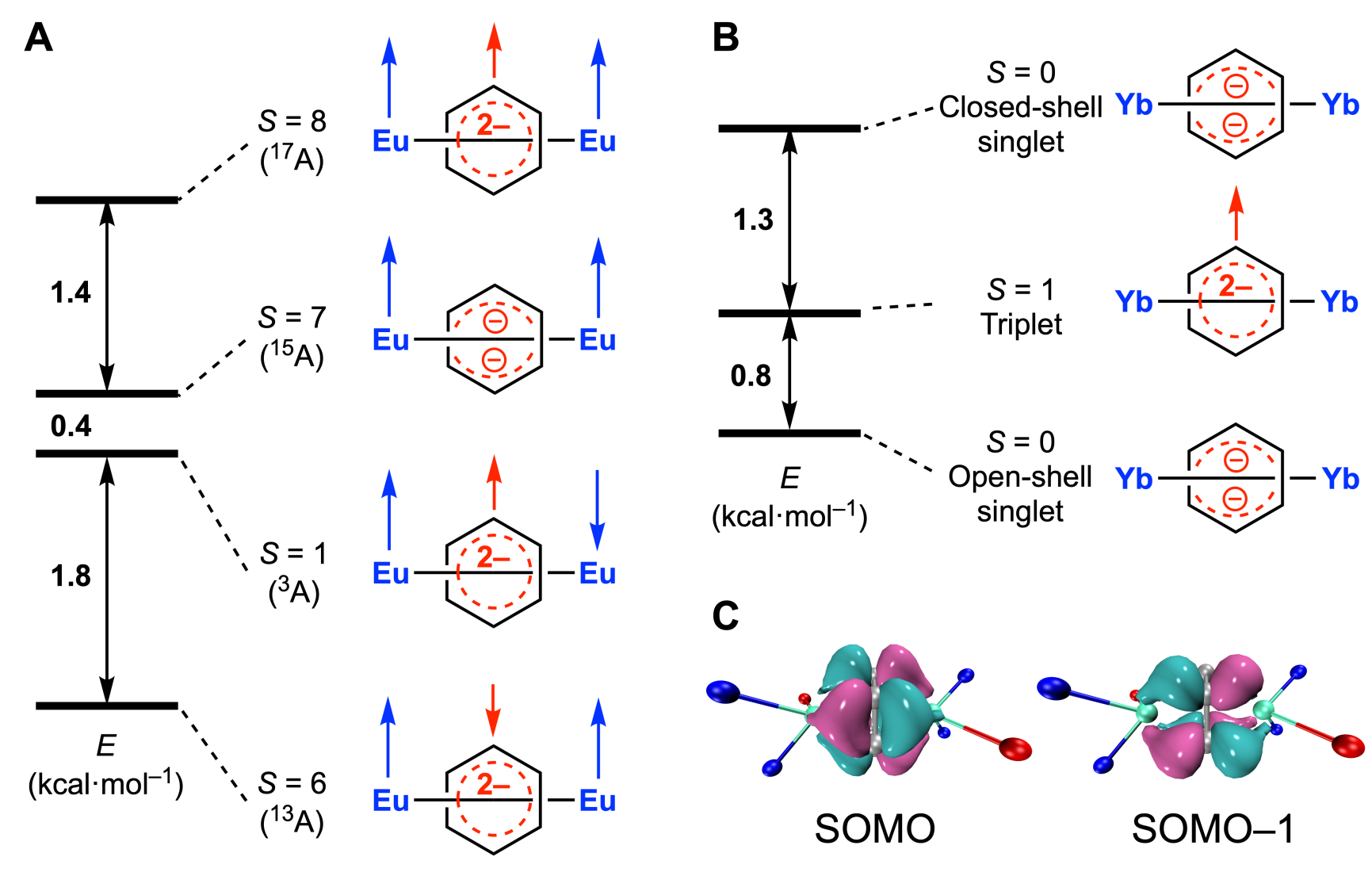
图4. 2-Yb,2-Eu和3-Eu的DFT计算结果(A)2-Eu不同自旋多重态的相对能量;(B)2-Yb不同自旋多重态的相对能量;(C)2-Eu(13A)的δ成键轨道
为了分析苯二负离子的芳香性,作者进行了Nuclear-independent chemical shifts (NICS)的计算。在2-Eu(13A)中,三重态苯二负离子的NICS(0/1)zz值为−35.7/−72.0 ppm,表明其具有很强的芳香性,符合Baird规则的预测。相反,在2-Yb(OST)中,单重态苯二负离子的NICS(0/1)zz值为+335.1/+300.0 ppm,表明其具有反芳香性,符合Hückel规则的判断。
综上,自Baird提出三重态芳香性概念半个世纪以后,具有三重态基态和Baird芳香性的苯二负离子终被合成。本工作利用d−π*δ成键与4f−rad自旋−自旋相互作用的协同效应克服了极易发生的Jahn−Teller畸变,实现了高度平面化的三重态苯二负离子的稳定。该结果为拓展具有基态Baird芳香性的分子提供了指引,并例证了利用稀土离子进行自旋调制的有效性,为开发基于自旋调控的新型功能材料和器件奠定了基础。
该论文的共同第一作者为77779193永利集团已毕业博士生王怡博士和孙荣博士,黄闻亮长聘副教授和王炳武研究员为共同通讯作者,梁杰锋博士、张玉柔、谈博文、邓翀博士、王一涵和高松教授为共同作者,杨逸鹄为计算部分提供了协助。该研究得到了国家自然科学基金、国家重点研发计划、永利集团、北京分子科学国家研究中心等的资助,并得到了永利集团分析测试中心、永利集团高性能计算平台等的支持。
原文链接:https://pubs.acs.org/doi/10.1021/jacs.4c17459

 北大化学微信
北大化学微信